
This page briefly describes the use of HeliQuest webserver. You can go to:
Procedure details
Calculation of the mean hydrophobicity
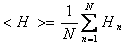
N is the sequence length and Hn is the hydrophobicity of the nth amino acid in the sequence, according to its octanol/water partition (Fauchère, J., and Pliska, V. 1983. Hydrophobic parameters {pi} of amino-acid side chains from the partitioning of N-acetyl-amino-acid amides. Eur. J. Med. Chem. 8: 369–375)
| Ala: | 0.310 | Arg: | -1.010 | Asn: | -0.600 |
| Asp: | -0.770 | Cys: | 1.540 | Gln: | -0.220 |
| Glu: | -0.640 | Gly: | 0.000 | His: | 0.130 |
| Ile | 1.800 | Leu: | 1.700 | Lys: | -0.990 |
| Met: | 1.230 | Phe: | 1.790 | Pro: | 0.720 |
| Ser: | -0.040 | Thr: | 0.260 | Trp: | 2.250 |
| Tyr: | 0.960 | Val: | 1.220 |
The <H> value ranges from -1.01 to 2.25
Calculation of the mean amphipathic moment <µH>
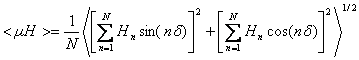
N is the sequence length, Hn is the hydrophobicity of the nth amino acid in the sequence and nδ is the angle separating side chains along the backbone with δ=100° for an alpha helix (Eisenberg, D., Weiss, R.M., and Terwilliger, T.C. 1982. The helical hydrophobic moment: a measure of the amphiphilicity of a helix. Nature 299: 371-374.) The length and the direction of the <µH> vector depend on the hydrophobicity and the position of the side chain along the helix axis. A large <µH> value means that the helix is amphipathic perpendicular to its axis.
The <µH> value ranges from to 0 to 3.26
Calculation of the net charge z
HeliQuest calculates the net charge at pH=7.4, considering that His is neutral and that the N-terminal amino group and the C-terminal carboxy group of the sequence are uncharged.
| Lys : | + 1 | Arg : | + 1 |
| Glu : | - 1 | Asp : | - 1 |
Analysis
Analysis form:
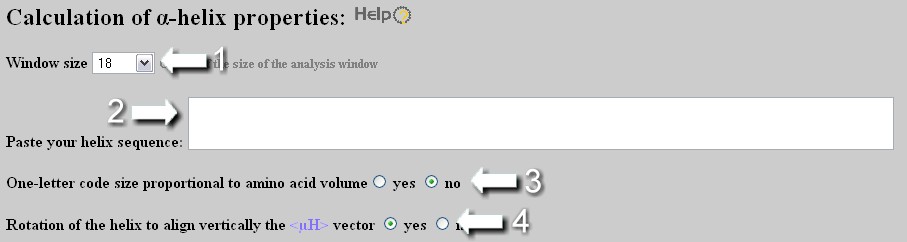
User (1) selects the size of the analysis window (FULL option corresponds to an analysis window of the size of the sequence with a limitation of 54 residues, i.e. three repeats of a complete helical wheel of 18 amino acids) and (2) enters the sequence. Important: select an analysis windows of 18 amino acids in order to access to the screening module from the results page. User can customize the helical wheel representation by representing the residues in function of their volume (3) and by rotating the helix in order to place downward its hydrophobic face, if existing (4).
Analysis results:
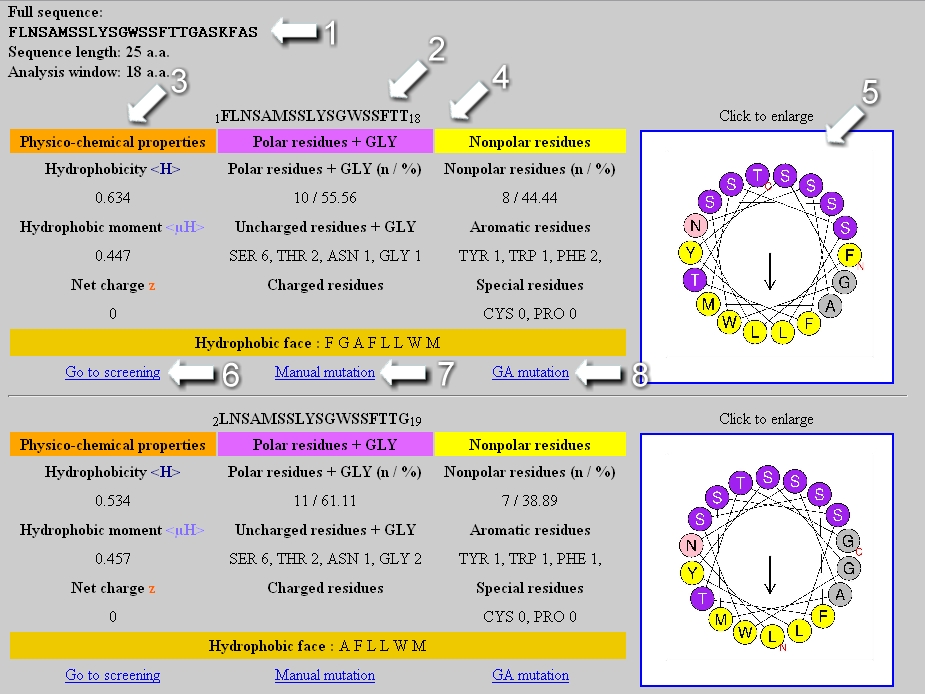
At the top of the page, the analyzed sequence as well as its length and the size of the analysis window are indicated (1). Several tables are generated if the analysis window is smaller than the sequence length (for example, the two first tables appear above in the Figure). Each table is associated with the sequence of the considered segment (2) and its helical wheel representation (5), that is downloadable as a jpeg format file. In the first column of the table, the physicochemical properties <H>, <µH> and z are reported (3). The second and third column correspond respectively to statistic on the polar and nonpolar residues present in the segment (4). If the analysis window is of 18 amino acids, a link to the screening module appears at the bottom of the table (6) as well as links to mutate the segment manually or automatically by genetic algorithm (7,8). At the bottom of the page, a downloadable graphic file represents the <H> and <µH> values of each segment in function of its position in the analyzed sequence.
Database screening
To use this module, one must select:
Eventually, user must indicate if the sequence can contain Pro at the very beginning or/and end of the sequence (among the first and/or last three residues) and/or Cys
Amphipathic helix identification algorithm.
To refine the identification of well-defined amphipathic helices, one can select Yes in the 'Geometric rules' box:
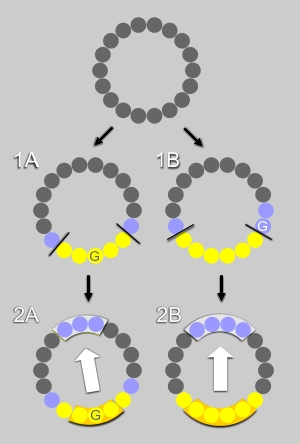
First step : the algorithm examines whether a segment contains an uninterrupted hydrophobic face, that is it contains at least 5 hydrophobic residues (Ala,Leu,Ile,Val,Met,Pro,Phe,Trp,Tyr) that are adjacent when represented on a helical wheel. For example, in Fig. 1A and 1B, the hydrophobic face contains respectively 5 or 6 residues (in yellow). The polar residues (in blue) at the edge of the hydrophobic face are recognized by the algorithm. Glycine represents an exception as it is the smallest residue; it is neutral regarding the hydrophobic scale and very flexible. If a glycine is flanked by two hydrophobic residues, it is considered as hydrophobic and counts as one of the residues of the hydrophobic face (Fig.1A). If a glycine is localized between a hydrophobic and a polar residue, it is considered as a polar residue at the edge of the hydrophobic face (Fig.1B).
Second step : if a hydrophobic face exists, the procedure examines whether the facing residues are polar or poorly hydrophobic (Ala,Asp,Glu,Gly,His,Lys,Asn,Gln,Arg,Ser,Thr). Depending on the number of residue in the hydrophobic face (odd or even) three or four residues were considered, (Fig. 2A and Fig. 2B, respectively).
DATABASE choice
SWISSPROT Databases The User selects the type of SWISSPROT databases .
| Organism | Number of sequences | Date |
| Human | 20367 | 2020:02:17 |
| Mouse | 17027 | 2020:02:17 |
| Rat | 8085 | 2020:02:17 |
| Yeast/font> | 20199 | 2020:02:17 |
For screening SWISSPROT database, it is possible to apply a filter termed Blacklist by cliking Yes to eliminate from the final selection the proteins for which a precise functional information is lacking (whose description contains terms of our blacklist: Hypothetical, Putative, Probable).
Personal Database The User selects the
location of its own personal database.
The personal database must be in a FASTA format.
A sequence in FASTA format begins with a single-line description,
followed by lines of sequence data. The description line is
distinguished from the sequence data by a greater-than (">")
symbol in the first column. The name following the ">" symbol is
the identifier of the sequence, and the rest of the line is the
description (both are optional). There should be no space between the
">" and the first letter of the identifier.The sequence ends if
another line starting with a ">" appears; this indicates the
start of another sequence.
Example of personal database: download example
> test sequence 1 personal databases protein y
AKLHTGFREDCVLKILKILKIKLKI.........
> sequence 2 protein x
MLHGFERCSQAGGHILMPVHIRTFDESESSEDP......
Important:, the E-mail address must be valid to receive the results of the screening by E-mail.
Decision Tree.
HeliQuest helps users
to analyze screening results. In particular, since HELIQUEST considers
any
This decision tree orders segments positive for the screening in six defined classes (TM segment, Helix, Helix/Coil , Possible Lipid-Binding Helix, Lipid-Binding Helix , High propensity in b-sheet).
Server first runs TMHMM to detect transmembrane segments that are helices whose specific properties permit prediction. Thereafter, for sorted segments that are not predicted as transmembrane, server runs PSIPRED to calculate their propensity to be helical, in b-sheets or in random coil in the context of the protein. Segments are also examined to see if their physicochemical properties correspond to those of known lipid-binding amphipathic helices. This prediction is based on a discrimant analysis that we performed on 48 amphipathic helices described unambiguously in literature as interacting (class 1) or not (class 2) with the surface of large, negatively-charged liposomes.
List of amphipathic segment and details
of the
analysis are given in Appendix.
Our analysis indicated that combining z and
<µH> values permits to define
a discriminant axis (with variable D) that segregates segments that
have no
lipid-binding ability from those that bind to biomimetic membranes.
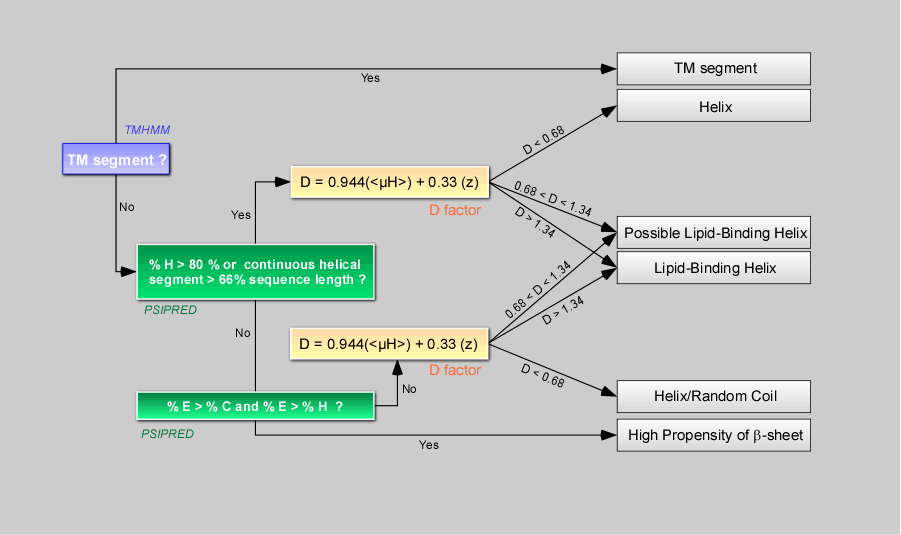
Note 1 : The discriminant factor D, based on
the
net charge z , is very sensitive to the number of charged residues in a
sequence
(discrete number)– thus, to avoid any boundary effect
(threshold value D=1.01) in
the classification of segment as lipid-binding or non lipid-binding, we created an intermediate
class termed “Possible
Lipid Binding Helix”. The D values of 0.68 and 1.34
corresponds respectively to
D=1.01 - 0.33*z and D=1.01 + 0.33*z with z=1.
Note 2: Segments with a high propensity of b-sheet are not further
considered in term of lipid-binding helices and could be considered as
non
relevant.
Note 3: Known lipid-binding segments are often
predicted either as fully helical or as a mix of random coil and
helical
structure. Thus, regardless of its level of helical propensity, a
segment
associated with D>1.33 or with a D value between
0.68 and 1.33 is
classified respectively as a Lipid-Binding Helix or a Possible
Lipid-Binding
Helix
The tests of HeliQuest are given in appendix.
Output file:
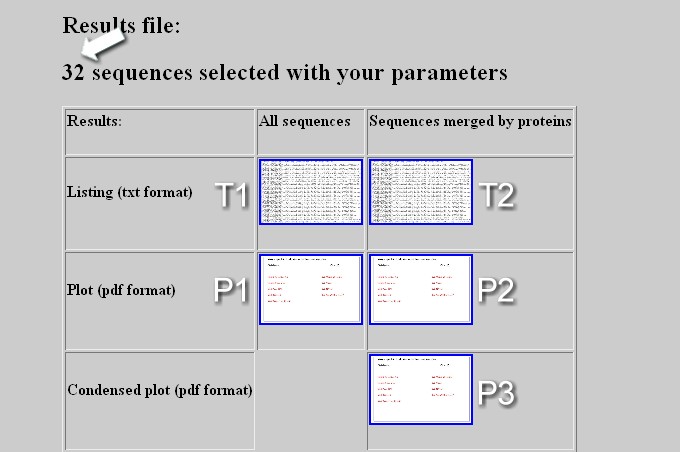
The output webpage displays the total number of sequence positive for the screening (arrow) and contains a table with links to download several types of file:
Sequence generation by Genetic Algorithm
This module allows applying three main strategies to mutate a helix and two others to design from scratch a novel helix.
The final sequence is provided with a GA score between 0 and 1.The optimal solution corresponds to a score of 1.
Mutation - Strategy 1
To only change the hydrophobic moment <µH> of a helix without modifying its amino acid composition. This could help design a set of analogues to see experimentally how the amphipathicity of a helix, independently of others parameters, influences its function.

On a reference sequence, the GA applies an unlimited number of mutation/permutation to reach the required <µH> value with the best GA score. If the <H> or z values are modified, the algorithm does not take into account these new values except if the 'Permutation only' option is deactivated (see Strategy 2)
Mutation - Strategy 2
To change the hydrophobicity <H> , the hydrophobic moment <µH> or the net charge z of a helix independently or in a combined manner with a maximal allowed number of mutation. As any change of hydrophobicity or net charge requires modifications of amino acid composition, one must select no in the 'Permutation only' box. User selects the maximal number of mutation allowed to reach the new <H>, <µH> and z values.
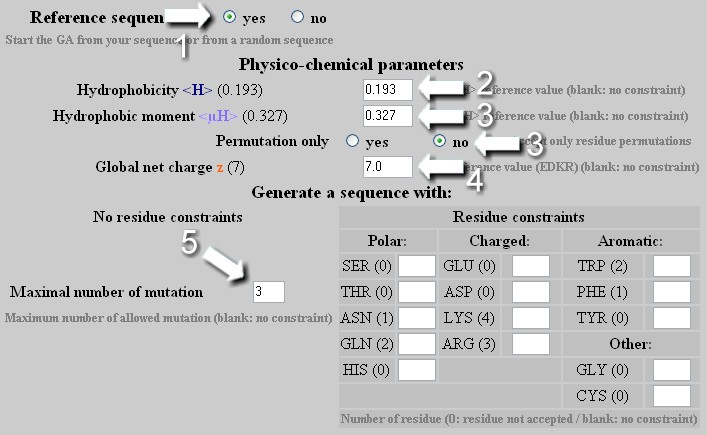
From a reference sequence, the GA will attempt first to find a solution with a number of mutation lower than that indicated by the user. If this solution exists, it will be the last one displayed by the GA. If not, the result obtained with the maximal number of allowed mutations will be shown even if no optimal sequence is found (no convergence). User can indeed increase the number of allowed mutation to obtain a better result or allow an unlimited number of mutation.
Mutation - Strategy 3
To change the hydrophobicity and the hydrophobic moment of a helix independently or in a combined manner with a specific composition of amino acid. Warning: as soon as the amino acids table is edited :
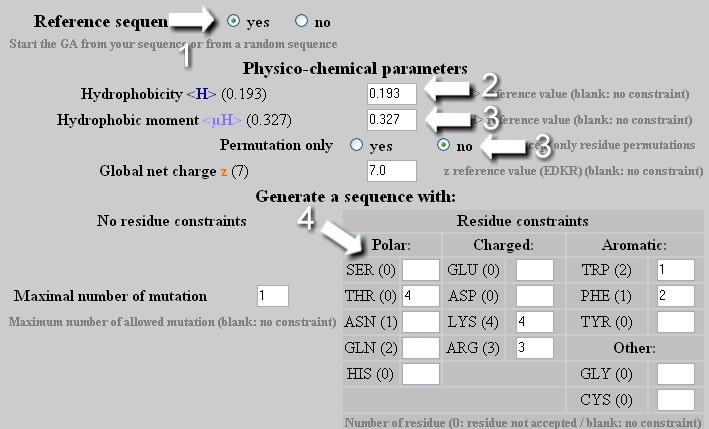
From a reference sequence, the GA will attempt to reach the desired <H>, <µH> and amino acid composition with the best GA score.
Design – Strategy 1
To create a helix with precise <H>, <µH> and z values but with a sequence unrelated to the reference helix.

The GA applies on a random sequence, an unlimited number of mutation/permutation to reach the desired <H>, <µH> and z values.
Design – Strategy 2
To create a helix with precise <H>, <µH> and z values and a specific content of amino acids determined by the user. Warning: as soon as the amino acids table is edited : the z value is no longer a constraint in the mutation process. The only way to precisely define z is to indicate the number of Glu, Asp, Arg and Lys in the amino acids table.

The GA applies on a random sequence an unlimited number of mutation/permutation to reach the required <H>, <µH>, z values and amino acids composition.
Automatic sequence mutation by a genetic algorithm (GA)
Genetic algorithms are artificial intelligence algorithms developed to solve combinatorial optimization problems for which the exact solution is intractable. Briefly, a genetic algorithm mimics some of the major characteristics of Darwinian evolution. The GA-based module generates novel peptides possessing properties that are precisely defined by the user. Starting from an initial sequence (from the reference sequence or from a sequence randomly generated), the GA applies crossover and mutation operators to generate a population of new peptides. Each peptide is evaluated by using a fitness function. The fitness function is a user-defined combination of various features: <H>, <µH>, z and the amino acid composition. The fitter the peptide, the more likely it would be selected to produce an offspring at the next generation. This selective pressure acts as a natural selection that favors the better solutions. The population of peptides evolves step by step toward an optimal solution. The best peptide is proposed.
Here, the algorithm works with a population of 40 peptides that are evolved during 2000 generations. Each new peptide is created by applying:
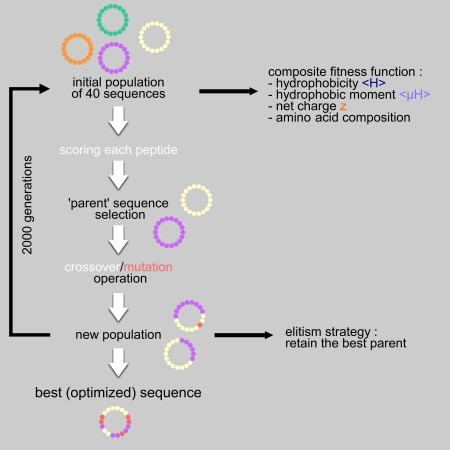
Thus, a new sequence can derive from an unmodified or a mutated combination of two parents or from a unique parent that is mutated or not. Mutations correspond either to substitutions or to permutations of amino acid in case of a fixed length peptide. Parent selection and operators process are applied 40 times.
The old generation is totally replaced by the new one but the worst peptide of the new population is systematically replaced by the best peptide of the precedent generation. This is called an elitism strategy that ensures that the best solution is never lost.
Manual mutation
The user can edit, shorten or lengthen the sequence – The number of each amino acid on the helical wheel representation is indicated to facilitate the edition of the sequence. Once edited, the sequence is submitted to the analysis module by clicking on the “Process” button
Mutation results:
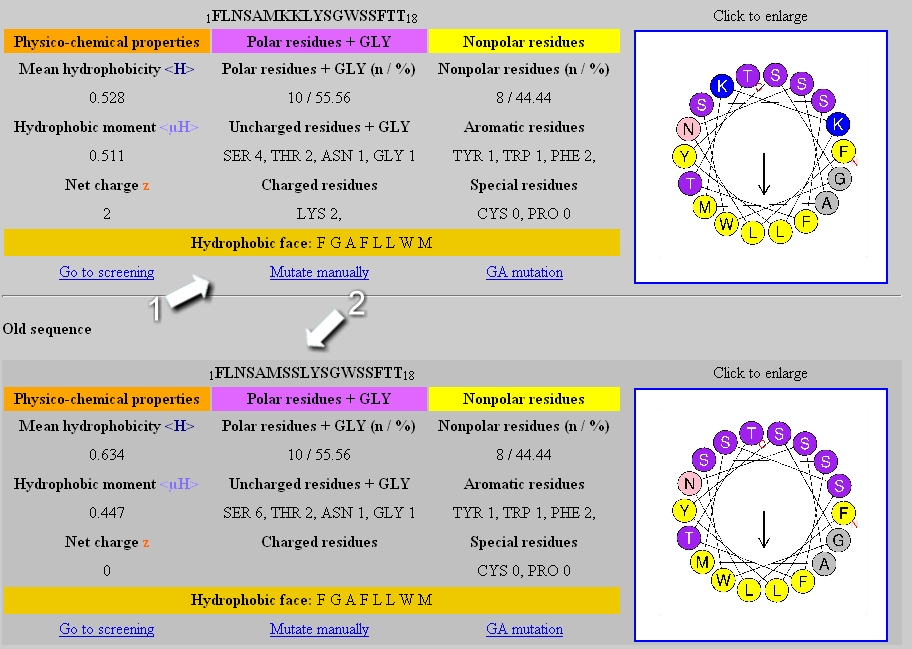
The table of the new (1) and the previous sequence (2) are displayed in the result page.